
![[headshot]](https://cdn.prod.website-files.com/68e9289a324a09160f2cdcc4/69826868dfda88e10069cb44_Betsy%20(2).png)
.png)
Subscribe our newsletter to receive the latest blogs, case studies and publications.
SubscribeEarly RWE planning gives teams the evidence needed to reduce uncertainty, defend value, and support coverage at launch. When planning begins after approval, the landscape is set and there is limited ability to change the access trajectory.
Exhibit 1: Five Disciplines of Access-Ready RWE
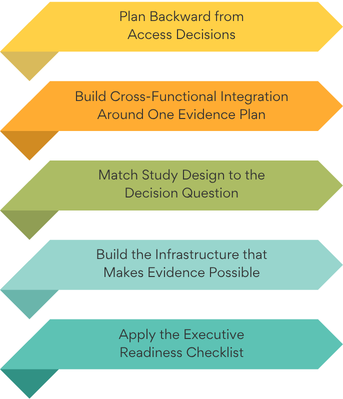
Regulators, payers, and HTA bodies increasingly expect real-world evidence. Infrastructure to generate credible evidence must be built long they ask for it. Many life sciences companies still treat RWE as a post-launch activity. That reactive model leaves teams managing access decisions after payer expectations have already formed.
Planning discipline is what separates RWE programs that change access outcomes from those that only generate publications.
This paper outlines a practical framework for building RWE programs around access decisions, decision-maker standards, and lifecycle value. The goal is simple: help HEOR, Market Access, R&D, Medical Affairs, Marketing, and Commercial leaders treat RWE as a strategic capability, not a late-stage publication activity.
Life sciences companies often build their RWE programs around a logical but limiting sequence: complete clinical development, earn regulatory approval, and then commission observational studies to satisfy post-marketing commitments, respond to payer evidence requests, and fill publication pipelines. Under this model, RWE begins after pivotal decisions have been made.
By the time most observational programs produce results, the commercial environment that shapes access is formed. Payers have set formulary tiers, written utilization management criteria, and established reimbursement restrictions. Payer expectations about comparative value have crystallized around the evidence available when those decisions were made.
Exhibit 2: From Reactive to Proactive Strategy
.png)
Evidence planned early becomes a commercial asset. Reactive evidence generation creates cost, delay, and uncertain return.
Changing those impressions after the fact is substantially harder than shaping key factors in advance. Formulary committees may be reluctant to remove step therapy requirements on the basis of new evidence alone, particularly when utilization controls have already been established.
Budget impact assumptions embedded in payer models persist through subsequent review cycles. Across high-scrutiny launches, companies that bring decision-grade evidence to key access reviews are better positioned for favorable initial coverage than companies that arrive with gaps and promises of future corrective programs.
Medicare’s Coverage with Evidence Development program provides a limited pathway to coverage when evidence questions remain unresolved at launch. Coverage may be available through an approved post-launch trial or registry, but this pathway is uncommon. More often, insufficient evidence leads to delayed, restricted, or denied access.
Except in special cases, such as early access programs, product RWE cannot be generated before launch. However, the foundation for strong comparative product RWE can—and should—be built during clinical development. Teams can generate landscape RWE to clarify the disease burden, treatment patterns, unmet needs, and real-world context for value. These efforts make RWE planning an essential part of development, not a post-launch activity. The next section examines these two types of RWE.
One of the most persistent planning errors in evidence strategy is treating RWE as a single category. In practice, the type of evidence a program can generate, the questions it answers, and the audiences it serves all depend on where the product sits in its development and commercial lifecycle. Treating pre- and post-launch evidence activities as the same discipline leads to predictable failures: pre-launch teams skip foundational work that would have strengthened launch positioning, and post-launch teams scramble to build infrastructure that should have been operational at launch. The most useful distinction for planning purposes is to consider landscape RWE versus product RWE, often requiring different data sources, study designs, and governance structures.
Exhibit 3: Landscape RWE vs. Product RWE
.png)
Manufacturers that define the disease baseline with their own real-world evidence control the comparison.
Although the product is not yet approved for commercial sale, teams can build the infrastructure that makes RWE possible at launch. This includes data partnerships with health systems and payers, registry design and governance, outcome measurement specs, and burden of illness evidence that anchors the value argument.
Published disease literature can lag current practice. Relying on it exclusively creates commercial risk in therapeutic areas where the standard of care is changing. When a product enters a market with evidence that is several years old, the launch value story rests on an outdated foundation. Real-world database analyses, claims studies, natural history data, and registry evidence can provide a more current picture of the clinical and competitive environment at launch. Landscape RWE establishes the empirical baseline against which the product’s value will be judged.
Landscape RWE also serves a strategic function beyond evidence generation itself. Burden of illness data, treatment pattern analyses, and care gap quantification inform endpoint selection, positioning, market segmentation, and value argument structure before companies engage external audiences. Companies that build this foundation early arrive at payer pre-engagement conversations with evidence rather than projections. That distinction can materially affect the outcome of future negotiations.
Companies often fund post-launch RWE, yet underinvest in pre-launch RWE.
A review of patient journey publications (Cribbs et al. 2025) found that 90% of studies captured healthcare utilization metrics. Yet, fewer than half captured essential decision-maker factors, such as treatment experience and disease management outcomes, and more than 75% omitted economic cost or productivity outcomes.
This imbalance limits the value of RWE for access planning. Utilization data can show how patients interact with the healthcare system, but it may not capture the outcomes decision-makers need to understand unmet need or assess comparative value.
Effective RWE planning must prioritize data sources and outcomes based on decision-maker relevance. Landscape RWE should establish the burden of disease, gaps in current care, and unmet needs. Product RWE should build comparative evidence base needed to demonstrate value after launch.
The framework below maps ten evidence uses across the product lifecycle. Pre-launch evidence builds the access foundation by defining unmet need, current care, and decision-maker priorities. Post-launch evidence sustains, defends, and extends product value through ongoing demonstration, access renegotiation, and lifecycle expansion.
Exhibit 4: Pre-Launch RWE
.png)
The commercial logic is straightforward: payers and HTA bodies evaluate products against the current standard of care. Teams that define that baseline with their own real-world evidence are better positioned to shape the comparison.
Early investment creates an empirically grounded narrative about the problem the product solves: who the patients are, how current treatments perform, what management paradigms cost the system, and where the care pathway breaks down. While this foundation does not guarantee favorable coverage, it does reduce the evidence gaps that often lead to restrictive payer coverage conditions.
Exhibit 5: Post-Launch RWE
.png)
Regulatory strategy affects access planning because it helps define the evidentiary baseline payers will later assess. Aligning regulatory and access evidence strategies can preserve budget, reduce duplication, and avoid parallel evidence programs.
This alignment is increasingly important because regulatory and payers and HTA bodies can collaborate directly and early (e.g., joint review). Additionally, each publishes their own frameworks and methodological standards for how RWE should be generated, evaluated, and applied.
Payer systems and HTA frameworks differ by market, but their core access questions are consistent: What problem does the product solve, for whom, compared with what, at what cost, and with what degree of confidence? RWE programs structured around those questions create the clearest path to decision-grade evidence.
Exhibit 6: Five Questions Decision-Makers Ask
.png)
As regulators and payers review more RWE submissions, their methodological expectations continue to rise. Submissions that fall short can undermine credibility with reviewers who are increasingly able to identify weaknesses in study design, data quality, and reporting. In some cases, weak RWE may create more risk than submitting no evidence at all.
Exhibit 7: Standards for Decision Grade Evidence
.png)
RWE requirements vary across payer and HTA systems, but several structural patterns hold across major markets. HEOR and Market Access teams need to understand these patterns when building global evidence programs.
Exhibit 8: Payer and HTA Expectations by Body
.png)
Patients can reach treatment sooner when evidence is planned before decision-makers ask for it.
The case for early, structured RWE investment is clear. NHS patient access data, FDA approval records, and peer-reviewed analyses of label expansion patterns show that RWE performs best when treated as a planning discipline. Across these examples, stronger access outcomes appear linked to earlier, more specific evidence design.
The NHS Cancer Drugs Fund in England demonstrates how structured RWE planning can support patient access. Since the program was redesigned in 2016, approximately 102,114 patients have received cancer treatments through the Fund, spanning 279 indications and 116 drugs. (NHS 2024)
More than 63,000 patients accessed treatments through managed access arrangements while evidence collection was ongoing. An additional 23,500 received interim access because a credible, pre-specified evidence plan gave NICE’s appraisal committee confidence that remaining uncertainty would be actively addressed.
The lesson is that structured planning, credible governance, and decision-linked evidence generation can make access possible before final evidence is available.
Longitudinal real-world data can reveal patient segments that benefit from a therapy beyond its original label. When those signals are credible, they can inform evidence generation for label expansion and help extend access to patients who would otherwise remain outside the approved population.
Palbociclib’s 2019 supplemental approval for male patients with hormone receptor-positive, HER2-negative advanced or metastatic breast cancer illustrates how RWE can support access for populations underrepresented in pivotal trials. Because breast cancer is rare in men, the clinical trial evidence base was limited. Real-world data helped show that men were receiving and responding to palbociclib in routine practice, contributing to the evidence package for label expansion.
This pattern is becoming more common. A 2024 review of FDA supplemental approvals from January 2022 through May 2024 found that RWE contributed, or likely contributed, to 25.2% of label expansions. (Cohen et al. 2024) That figure may understate RWE’s role because FDA public documentation does not always disclose which data sources influenced the final decision.
RWE can generate lifecycle management insights for both strategic brands and the broader portfolio. Regular reviews of real-world data can surface unanticipated risks or benefits, identify early trends, reveal shifts in care patterns, and support competitive threat mitigation.
Established RWE programs also provide a strategic buffer when assets face unexpected challenges. When concerns emerge around a perceived class effect, suspected safety signal, or competitive threat, teams with mature RWE infrastructure are better positioned to mobilize relevant data quickly. That evidence-based response may help address uncertainty and reduce the risk of restrictions driven by incomplete information.
These examples create a strong investment rationale. Real-world data can contribute meaningfully to lifecycle strategy, portfolio protection, and global revenue potential. For that reason, RWE infrastructure should be built early in the commercial planning cycle. With the right planning, RWE becomes a strategic asset in its own right: a source of insight and evidence that protects the portfolio, supports expansion, and helps mitigate clinical and commercial risk.
An evidence portfolio can be individually reasonable and still collectively misaligned. That is one of the most expensive failures in commercial planning.
Many RWE programs fail structurally because they plan forward from study initiation instead of backward from the anticipated access decision.
Backward planning reverses the sequence. Teams first identify the likely timing of HTA submissions, formulary reviews, managed access renewals, contracting milestones, and reimbursement reassessments that will determine access. They then specify the evidence each decision requires and calculate the latest start date that still allows results to be available before the decision occurs.
The exercise consistently reveals that critical evidence activities must begin earlier than teams expect, particularly when studies depend on data partnerships, registry governance, or longitudinal follow-up.
The output is a decision-linked evidence map: each access event is tied to a submission deadline, required deliverable, accountable owner, and initiation date.
Evidence programs usually fail at the boundaries between functions. Market Access builds arguments from the evidence available. HEOR designs studies around scientific priorities. Medical Affairs manages publications and scientific exchange. Commercial builds value narratives from clinical data. Each workstream may be sound in isolation, but without a shared organizing framework, the portfolio becomes fragmented and misaligned with the access objectives it was meant to serve.
Effective evidence programs start with an integrated plan. The plan defines the proof domains required for launch and lifecycle management, including burden of illness, treatment patterns, comparative effectiveness, persistence and adherence, budget impact, and long-term safety and durability. Each activity is then tied to a named decision milestone and a primary stakeholder audience.
Designing a study around a relevant question sounds obvious. Yet many RWE studies start with the available dataset rather than the decision the evidence must support.
Payer claims are often the default choice under time pressure, but they were not designed for research. They can support credible evidence only with careful curation, fit-for-purpose assessment, and transparent methods. Payer and HTA reviewers can recognize studies designed for convenience rather than decision relevance, and they discount the evidence accordingly.
For causal questions, target trial emulation offers a defensible observational approach because it makes the comparison explicit before analysis begins. Investigators define the randomized trial they would have run, then design the observational study to emulate it as closely as possible. That means specifying eligibility criteria, treatment strategies, time zero, outcomes, and follow-up before analyzing the data. NICE’s RWE framework recommends this approach for non-randomized comparative effectiveness studies. (NICE 2022)
Evidence infrastructure must exist before access depends on it. Registries, longitudinal follow-up programs, and structured data collection in clinical workflows cannot be commissioned at the moment a payer or regulator asks for evidence.
They require data access agreements, governance frameworks, workflow integration, fit-for-purpose registry design, quality oversight, validation, monitoring, and escalation processes for data quality issues. These components take longer to build than the window between a payer request and a formulary review. Reactive infrastructure planning therefore creates a predictable failure: the evidence is not ready when the decision will be made.
Leaders in Market Access, HEOR, Commercial, and Medical Affairs can use this checklist to assess whether their evidence program is ready to support access and lifecycle strategy. It focuses on the planning, governance, methodological, and infrastructure requirements that help prevent scientifically sound RWE programs from becoming commercially ineffective.
Exhibit 9: Executive Readiness Checklist
.png)
To turn RWE into access impact, we call on leaders across Market Access, HEOR, Commercial, and Medical Affairs to build RWE into access strategy.
Build RWE Into Development Strategy:
Evidence planning aligned with payer and HTA requirements should begin in Phase II planning. Endpoint selection, patient-reported outcome specification, real-world data partnerships, and study design choices can shape pivotal development and future access in ways that are difficult to correct later.
Strategic Question:
"How do we build the evidence foundation needed to establish burden, unmet need, and differentiated value before the market is defined without us?"
Scenario Plan Proactively:
Every major commercial decision carries an access policy dimension. MFN, IRA negotiation timelines, orphan exemption structures, coverage with evidence development requirements, and HTA joint clinical assessment frameworks all create strategic inflections with evidence implications that should be addressed before launch.
Strategic Question:
"How do we engage payers, HTA bodies, and policy stakeholders early enough to shape evidence expectations before access decisions are framed without our input?"
Demonstrate Ongoing Value:
Sustained reimbursement requires evolving evidence. Post-launch RWE programs that demonstrate long-term clinical and economic outcomes, support outcomes-based contract performance, and provide evidence for lifecycle expansion are now core commercial capabilities.
Strategic Question:
"Are we continuously generating and communicating comparative value in ways that protect price, preserve access, and support future growth?"
RWE planning that begins in clinical development creates a stronger launch position. By approval, disease burden can be characterized, the care pathway mapped, and data infrastructure prepared to generate real-world outcomes from day one of commercialization. That foundation does not appear at launch; it must be built years before approval.
Turning RWE into access impact requires one integrated evidence plan across HEOR, Market Access, Medical Affairs, Marketing, and Commercial. The plan should connect evidence activities to shared objectives, decision milestones, and access timelines. It should run backward from formulary reviews, HTA submissions, and managed access renewals, not forward from the research team’s preferred schedule. When a payer raises a question, the team should already know what evidence exists, where it sits, and how it will be used.
The access environment is becoming less forgiving. Payer scrutiny is rising, HTA expectations are tightening, and the window between regulatory approval and commercial traction is narrowing. RWE cannot remain a publication exercise or a post-launch contingency plan. The commercial advantage will belong to teams that build RWE before the market asks for it.
This guide is designed to be used independently of the paper, as a reference for teams working across evidence planning, payer submissions, and HTA dossier development. Definitions are written for functional leaders who work with evidence strategy but may not have a formal research methods background.
.png)
US Food and Drug Administration. Real-world evidence. FDA. Updated January 23, 2026. U.S. Food and Drug Administration. Real-World Evidence Program. https://www.fda.gov/science-research/science-and-research-special-topics/real-world-evidence
U.S. Food and Drug Administration. Guidance for Industry: Externally Controlled Trials to Support Drug and Biological Product Approval. https://www.fda.gov/media/164960/download
UK Medicines and Healthcare products Regulatory Agency. Guidance on the Use of Real-World Data in Clinical Studies to Support Regulatory Decisions. https://www.gov.uk/government/publications/mhra-guidance-on-the-use-of-real-world-data-in-clinical-studies-to-support-regulatory-decisions
Pharmaceuticals and Medical Devices Agency, Japan. Points to Consider When Registry Data Are Used for Partial Change Approval Applications or Revision of the Electronic Package Insert for Prescription Drugs. 2024.
CDA-AMC. Guidance for the Use of Real-World Evidence in Drug Submissions. Canadian Drug Agency. https://www.canada.ca/en/health-canada/services/drugs-health-products/drug-products/announcements/health-canada-position-guidance-reporting-real-world-evidence-supporting-decision-making.html
NICE. Real-World Evidence Framework. National Institute for Health and Care Excellence. https://www.nice.org.uk/corporate/ecd9/resources/nice-realworld-evidence-framework-pdf-1124020816837
Institut fur Qualitat und Wirtschaftlichkeit im Gesundheitswesen. General Methods, Version 8.0.
https://www.iqwig.de/en/about-us/methods/methods-paper/
Medical Affairs Professional Society. Strategic Integrated Evidence Generation Planning: Company and Non-Company Sponsored Research. 2025. https://medicalaffairs.org/wp-content/uploads/2025/02/EVIDENCE-GENERATION-Standards-and-Guidance-2-11-2025.pdf
Cribbs KA, Blackmore LTA, Banks AR, Kim DS, Lahue BJ. Capturing real-world rare disease patient journeys: are current methodologies sufficient for informed healthcare decisions? J Eval Clin Pract. 2025;31(2):e70010. doi:10.1111/jep.70010
NHS England. Cancer drugs fund. NHS England. https://www.england.nhs.uk/cancer/cdf/
Wedam S, Fashoyin-Aje L, Bloomquist E, et al. FDA approval summary: palbociclib for male patients with metastatic breast cancer. Clin Cancer Res. 2020;26(6):1208-1212. doi:10.1158/1078-0432.CCR-19-2580
Deng J, Girman C, Ritchey ME. Real-world evidence in FDA approvals for labeling expansion of small molecules and biologics. Ther Innov Regul Sci. 2025. doi:10.1007/s43441-025-00816-9
Vaghela S, Tanni KA, Banerjee G, Sikirica V. A systematic review of real-world evidence (RWE) supportive of new drug and biologic license application approvals in rare diseases. Orphanet J Rare Dis. 2024;19(1):117. doi:10.1186/s13023-024-03111-2
Centers for Medicare & Medicaid Services. Statement: broader Medicare coverage of Leqembi available following FDA traditional approval. CMS.gov. July 6, 2023. https://www.cms.gov/newsroom/press-releases/statement-broader-medicare-coverage-leqembi-available-following-fda-traditional-approval
US Food and Drug Administration. Advancing Real-World Evidence Program. FDA. https://www.fda.gov/drugs/development-resources/advancing-real-world-evidence-program
Alkemi. The "too late" test: a framework for evidence-ready launch planning. Alkemi Blog. June 18, 2025. https://www.alkemihealth.com/blog/the-too-late-test-a-framework-for-evidence-ready-launch-planning
Alkemi. Planning RWE after launch is like planning a clinical trial after enrollment. Alkemi Blog. November 25, 2025. https://www.alkemihealth.com/blog/planning-rwe-after-launch
Alkemi. Modern commercial strategy: why value & access must lead early. Alkemi Blog. February 11, 2026. https://www.alkemihealth.com/blog/modern-commercial-strategy
Alkemi. When RWE actually starts (hint: not at launch). Alkemi Blog. December 12, 2025. https://www.alkemihealth.com/blog/when-rwe-actually-starts
Alkemi. Outcomes-based contracting 2.0: why RWE readiness is becoming table stakes for access. Alkemi Blog. January 30, 2026. https://www.alkemihealth.com/blog/outcomes-based-contracting-2-0
Hernán MA, Robins JM. Using big data to emulate a target trial when a randomized trial is not available. Am J Epidemiol. 2016;183(8):758-764. doi:10.1093/aje/kwv254
Wang SV, Pottegård A, Crown W, et al. HARmonized protocol template to enhance reproducibility of hypothesis-evaluating real-world evidence studies on treatment effects: a good practices report of a joint ISPE/ISPOR task force. Value Health. 2022;25(10):1663-1672. doi:10.1016/j.jval.2022.09.001
Wang SV, Pinheiro S, Hua W, et al. STaRT-RWE: structured template for planning and reporting on the implementation of real world evidence studies. BMJ. 2021;372:m4856. doi:10.1136/bmj.m4856
Langan SM, Schmidt SA, Wing K, et al. The reporting of studies conducted using observational routinely collected health data statement for pharmacoepidemiology (RECORD-PE). BMJ. 2018;363:k3532. doi:10.1136/bmj.k3532
US Food and Drug Administration. Considerations for the design and conduct of externally controlled trials for drug and biological products: guidance for industry. FDA; February 2023. https://www.fda.gov/media/164960/download
Schedule a consultation below. Together, we’ll look at your situation and explore your value proposition. If we can help, we’ll explain how. If not, we’ll connect you with a partner who can.


